Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders
- PMID: 22048062
- PMCID: PMC4036520
- DOI: 10.1038/nrn3114
Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders
Abstract
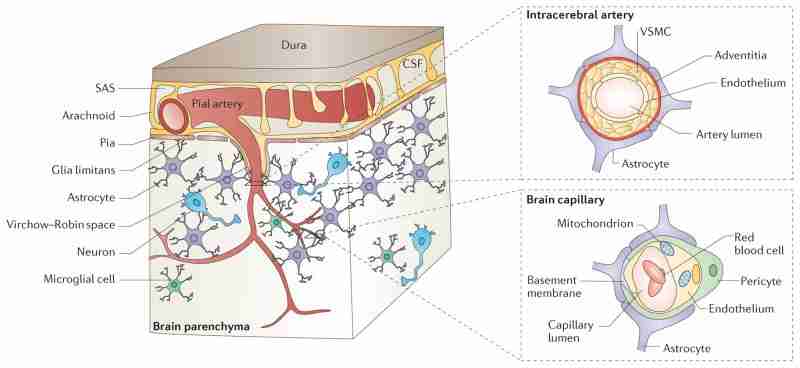
The neurovascular unit (NVU) comprises brain endothelial cells, pericytes or vascular smooth muscle cells, glia and neurons. The NVU controls blood-brain barrier (BBB) permeability and cerebral blood flow, and maintains the chemical composition of the neuronal 'milieu', which is required for proper functioning of neuronal circuits. Recent evidence indicates that BBB dysfunction is associated with the accumulation of several vasculotoxic and neurotoxic molecules within brain parenchyma, a reduction in cerebral blood flow, and hypoxia. Together, these vascular-derived insults might initiate and/or contribute to neuronal degeneration. This article examines mechanisms of BBB dysfunction in neurodegenerative disorders, notably Alzheimer's disease, and highlights therapeutic opportunities relating to these neurovascular deficits.
Conflict of interest statement
The author declares competing financial interests: see web version for details.
Figures
Similar articles
-
The blood-brain barrier in health and chronic neurodegenerative disorders.Neuron. 2008 Jan 24;57(2):178-201. doi: 10.1016/j.neuron.2008.01.003. Neuron. 2008. PMID: 18215617 Review.
-
Neurovascular mechanisms of Alzheimer's neurodegeneration.Trends Neurosci. 2005 Apr;28(4):202-8. doi: 10.1016/j.tins.2005.02.001. Trends Neurosci. 2005. PMID: 15808355 Review.
-
Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease.Brain Pathol. 2013 May;23(3):303-10. doi: 10.1111/bpa.12004. Epub 2012 Nov 28. Brain Pathol. 2013. PMID: 23126372 Free PMC article.
-
Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease.Acta Neuropathol. 2009 Jul;118(1):103-13. doi: 10.1007/s00401-009-0522-3. Epub 2009 Mar 25. Acta Neuropathol. 2009. PMID: 19319544 Free PMC article. Review.
-
The pericyte: a forgotten cell type with important implications for Alzheimer's disease?Brain Pathol. 2014 Jul;24(4):371-86. doi: 10.1111/bpa.12152. Brain Pathol. 2014. PMID: 24946075 Free PMC article. Review.
Cited by 709 articles
-
Morphological landscape of endothelial cell networks reveals a functional role of glutamate receptors in angiogenesis.Sci Rep. 2020 Aug 14;10(1):13829. doi: 10.1038/s41598-020-70440-0. Sci Rep. 2020. PMID: 32796870 Free PMC article.
-
Vascular Inflammation Is a Risk Factor Associated with Brain Atrophy and Disease Severity in Parkinson's Disease: A Case-Control Study.Oxid Med Cell Longev. 2020 Jul 14;2020:2591248. doi: 10.1155/2020/2591248. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 32733633 Free PMC article.
-
Acute effects of systemic inflammation upon the neuro-glial-vascular unit and cerebrovascular function.Brain Behav Immun Health. 2020 May;5:100074. doi: 10.1016/j.bbih.2020.100074. Brain Behav Immun Health. 2020. PMID: 32685933 Free PMC article.
-
Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease.Mol Neurodegener. 2020 Jul 16;15(1):40. doi: 10.1186/s13024-020-00391-7. Mol Neurodegener. 2020. PMID: 32677986 Free PMC article. Review.
-
Retinal and Brain Organoids: Bridging the Gap Between in vivo Physiology and in vitro Micro-Physiology for the Study of Alzheimer's Diseases.Front Neurosci. 2020 Jun 17;14:655. doi: 10.3389/fnins.2020.00655. eCollection 2020. Front Neurosci. 2020. PMID: 32625060 Free PMC article. Review.
Publication types
MeSH terms
Grant support
LinkOut - more resources
-
Full Text Sources
-
Other Literature Sources
-
Medical

{kind=link}