Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks
- PMID: 20581818
- PMCID: PMC3072750
- DOI: 10.1038/nn.2583
Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks
Abstract
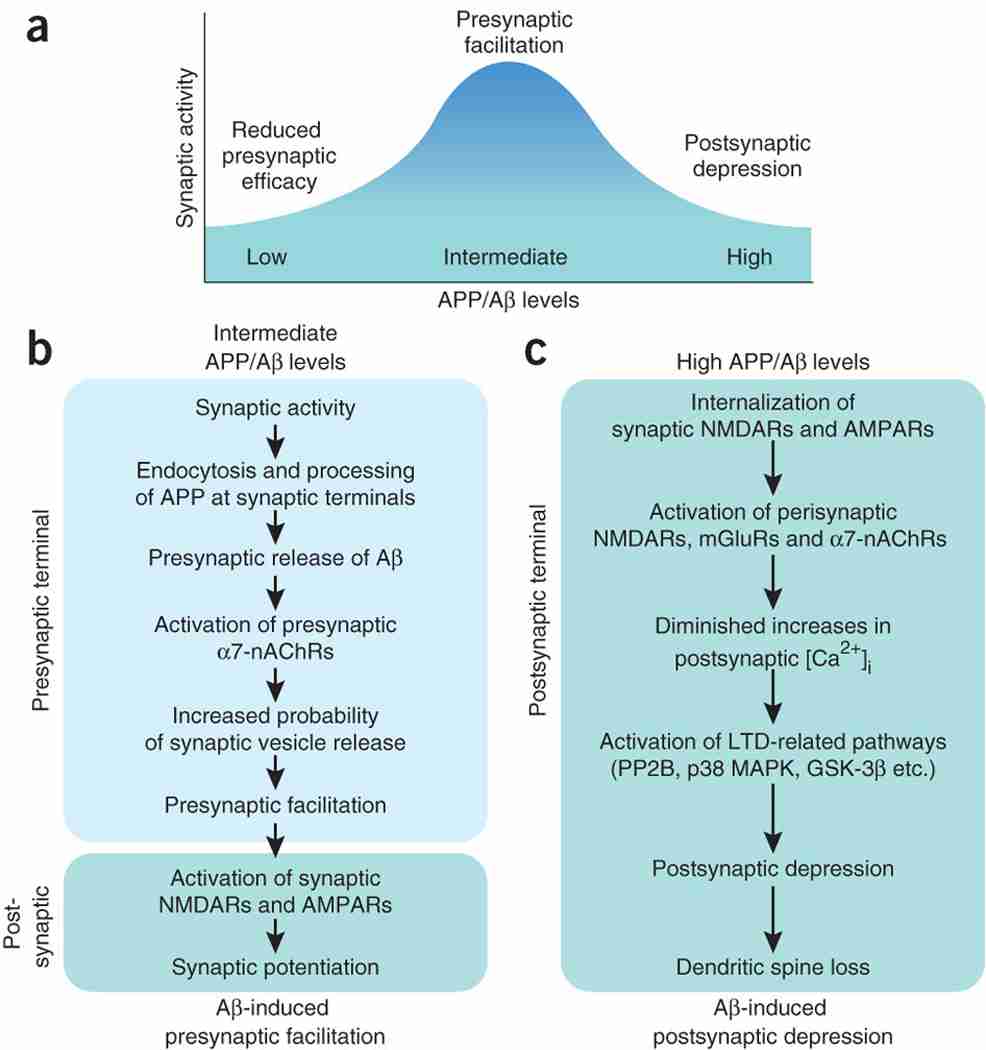
Alzheimer's disease is the most frequent neurodegenerative disorder and the most common cause of dementia in the elderly. Diverse lines of evidence suggest that amyloid-beta (Abeta) peptides have a causal role in its pathogenesis, but the underlying mechanisms remain uncertain. Here we discuss recent evidence that Abeta may be part of a mechanism controlling synaptic activity, acting as a positive regulator presynaptically and a negative regulator postsynaptically. The pathological accumulation of oligomeric Abeta assemblies depresses excitatory transmission at the synaptic level, but also triggers aberrant patterns of neuronal circuit activity and epileptiform discharges at the network level. Abeta-induced dysfunction of inhibitory interneurons likely increases synchrony among excitatory principal cells and contributes to the destabilization of neuronal networks. Strategies that block these Abeta effects may prevent cognitive decline in Alzheimer's disease. Potential obstacles and next steps toward this goal are discussed.
Figures
{kind=link}
Similar articles
-
Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease.Mol Neurodegener. 2014 Nov 14;9:48. doi: 10.1186/1750-1326-9-48. Mol Neurodegener. 2014. PMID: 25394486 Free PMC article. Review.
-
Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer's disease.J Neurochem. 2012 Jan;120 Suppl 1(Suppl 1):140-8. doi: 10.1111/j.1471-4159.2011.07506.x. Epub 2011 Nov 28. J Neurochem. 2012. PMID: 22122128 Free PMC article. Review.
-
Is Alzheimer's disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid.Rev Neurosci. 2009;20(1):1-12. doi: 10.1515/revneuro.2009.20.1.1. Rev Neurosci. 2009. PMID: 19526730 Review.
-
Alzheimer's disease-associated neurotoxic mechanisms and neuroprotective strategies.Curr Drug Targets CNS Neurol Disord. 2005 Aug;4(4):383-403. doi: 10.2174/1568007054546117. Curr Drug Targets CNS Neurol Disord. 2005. PMID: 16101556 Review.
-
Synapses and Alzheimer's disease.Cold Spring Harb Perspect Biol. 2012 May 1;4(5):a005777. doi: 10.1101/cshperspect.a005777. Cold Spring Harb Perspect Biol. 2012. PMID: 22491782 Free PMC article. Review.
Cited by 512 articles
-
GABAergic Inhibitory Interneuron Deficits in Alzheimer's Disease: Implications for Treatment.Front Neurosci. 2020 Jun 30;14:660. doi: 10.3389/fnins.2020.00660. eCollection 2020. Front Neurosci. 2020. PMID: 32714136 Free PMC article. Review.
-
Hyperexcitable Parvalbumin Interneurons Render Hippocampal Circuitry Vulnerable to Amyloid Beta.iScience. 2020 Jul 24;23(7):101271. doi: 10.1016/j.isci.2020.101271. Epub 2020 Jun 14. iScience. 2020. PMID: 32593000 Free PMC article.
-
Object-Based Analyses in FIJI/ImageJ to Measure Local RNA Translation Sites in Neurites in Response to Aβ1-42 Oligomers.Front Neurosci. 2020 Jun 3;14:547. doi: 10.3389/fnins.2020.00547. eCollection 2020. Front Neurosci. 2020. PMID: 32581689 Free PMC article.
-
A Defined and Scalable Peptide-Based Platform for the Generation of Human Pluripotent Stem Cell-Derived Astrocytes.ACS Biomater Sci Eng. 2020 Jun 8;6(6):3477-3490. doi: 10.1021/acsbiomaterials.0c00067. Epub 2020 May 6. ACS Biomater Sci Eng. 2020. PMID: 32550261 Free PMC article.
-
A human induced pluripotent stem cell-derived cortical neuron human-on-a chip system to study Aβ42 and tau-induced pathophysiological effects on long-term potentiation.Alzheimers Dement (N Y). 2020 May 27;6(1):e12029. doi: 10.1002/trc2.12029. eCollection 2020. Alzheimers Dement (N Y). 2020. PMID: 32490141 Free PMC article.
Publication types
MeSH terms
Substances
Grant support
LinkOut - more resources
-
Full Text Sources
-
Other Literature Sources
-
Medical